
![]()
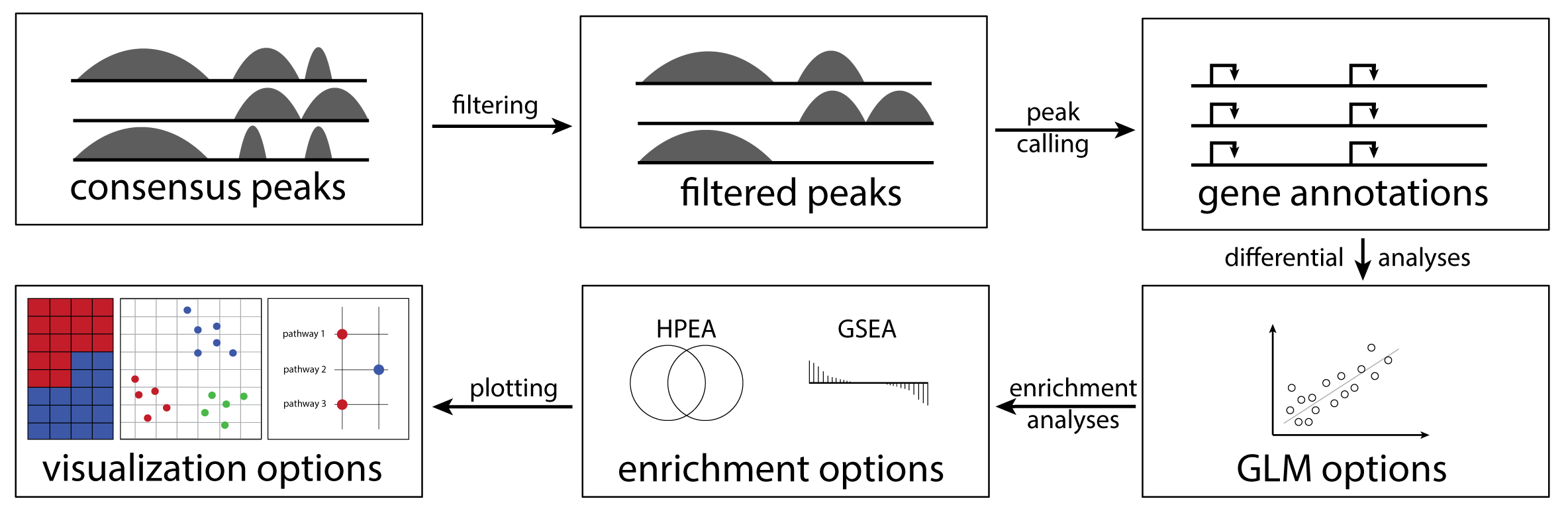
cinaR is a single wrapper function for end-to-end
computational analyses of bulk ATAC-seq (or RNA-seq) profiles. Starting
from a consensus peak file, it outputs differentially accessible peaks,
enrichment results, and provides users with various configurable
visualization options. For more details, please see the preprint.
# CRAN mirror
install.packages("cinaR")To get bug fix and use a feature from the development version:
# install.packages("devtools")
devtools::install_github("eonurk/cinaR")Sometimes bioconductor related packages may not be installed
automatically.
Therefore, you may need to install them manually:
BiocManager::install(c("ChIPseeker", "DESeq2", "edgeR", "fgsea","GenomicRanges", "limma", "preprocessCore", "sva", "TxDb.Hsapiens.UCSC.hg38.knownGene", "TxDb.Hsapiens.UCSC.hg19.knownGene", "TxDb.Mmusculus.UCSC.mm10.knownGene"))library(cinaR)
#> Checking for required Bioconductor packages...
#> All required Bioconductor packages are already installed.
# create contrast vector which will be compared.
contrasts<- c("B6", "B6", "B6", "B6", "B6", "NZO", "NZO", "NZO", "NZO", "NZO", "NZO",
"B6", "B6", "B6", "B6", "B6", "NZO", "NZO", "NZO", "NZO", "NZO", "NZO")
# If reference genome is not set hg38 will be used!
results <- cinaR(bed, contrasts, reference.genome = "mm10")
#> >> Experiment type: ATAC-Seq
#> >> Matrix is filtered!
#>
#> >> preparing features information... 2024-05-22 12:38:01
#> >> identifying nearest features... 2024-05-22 12:38:02
#> >> calculating distance from peak to TSS... 2024-05-22 12:38:02
#> >> assigning genomic annotation... 2024-05-22 12:38:02
#> >> assigning chromosome lengths 2024-05-22 12:38:11
#> >> done... 2024-05-22 12:38:11
#> >> Method: edgeR
#> FDR:0.05& abs(logFC)<0
#> >> Estimating dispersion...
#> >> Fitting GLM...
#> >> DA peaks are found!
#> >> No `geneset` is specified so immune modules (Chaussabel, 2008) will be used!
#> >> enrichment.method` is not selected. Hyper-geometric p-value (HPEA) will be used!
#> >> Mice gene symbols are converted to human symbols!
#> >> Enrichment results are ready...
#> >> Done!
pca_plot(results, contrasts, show.names = F)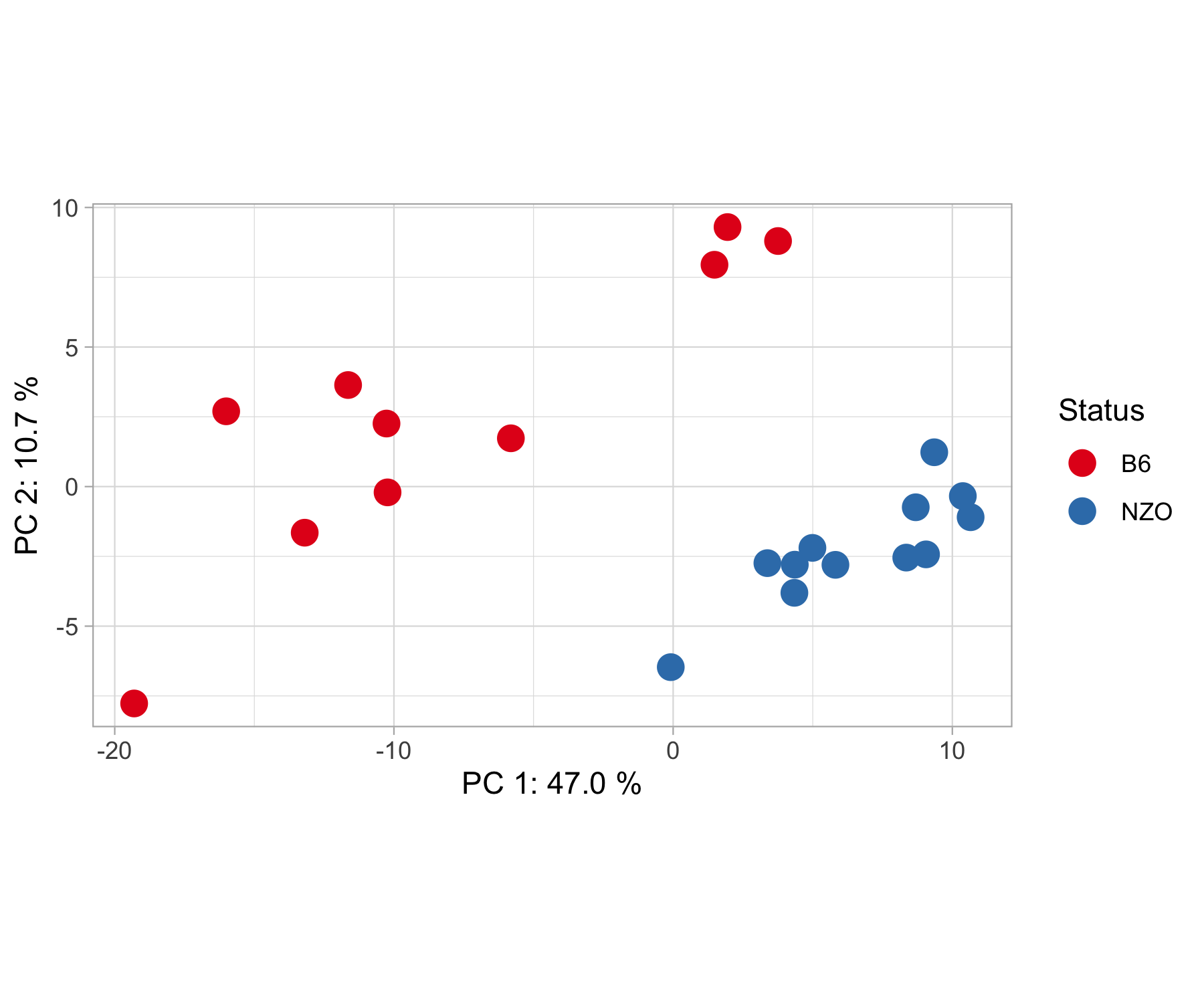
Use prep_scATAC_cinaR() to pseudobulk 10x scATAC
peak-by-cell matrices into a cinaR-ready consensus matrix.
This preserves biological replicates (sample-level) and avoids inflated
significance from per-cell testing.
# counts: peak-by-cell matrix (dense or dgCMatrix)
# meta: data.frame with rownames = cell barcodes
# meta must include biological replicate and condition columns
prep <- prep_scATAC_cinaR(counts, meta,
sample.col = "sample",
group.col = "group")
results <- cinaR(prep$bed, prep$contrasts, reference.genome = "hg38")Per-cell-type (sample × cluster) pseudobulk:
prep_list <- prep_scATAC_cinaR(counts, meta,
sample.col = "sample",
group.col = "group",
cluster.col = "celltype")
results_list <- lapply(prep_list, function(x) {
cinaR(x$bed, x$contrasts, reference.genome = "hg38")
})Seurat/Signac object:
prep <- prep_scATAC_seurat(seurat_obj,
sample.col = "sample",
group.col = "group",
assay = "peaks")
results <- cinaR(prep$bed, prep$contrasts, reference.genome = "hg38")If your peak IDs are not in chr:start-end,
chr_start_end, or chr-start-end format, pass a
peak.bed data.frame with CHR,
START, and STOP columns via
peak.bed = ....
For more details please go to our site from here!
@article {Karakaslar2021.03.05.434143,
author = {Karakaslar, E Onur and Ucar, Duygu},
title = {cinaR: A comprehensive R package for the differential analyses and
functional interpretation of ATAC-seq data},
year = {2021},
doi = {10.1101/2021.03.05.434143},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/10.1101/2021.03.05.434143v2},
journal = {bioRxiv}
}You can send pull requests to make your contributions.